
In normal cells, errors in DNA coding trigger cell cycle arrest in the G1/S or G2M phases and activate cell cycle associated checkpoint pathways that mediate stage dependent DNA repair. Once the damage has been repaired, arrested cells may either remain permanently arrested, re-enter the cell cycle or undergo apoptosis (if the damage is too severe). In cancer, the uncontrolled growth characteristic of the disease is associated with decreased time spent at DNA repair checkpoints, their complete bypass, poor or incomplete DNA repair and accelerated proliferation in the more aggressive forms. The search for improved cancer therapies often focuses on DNA repair checkpoint restoration, which can be studied by monitoring cell cycle progression and timing.
Aside from immunophenotyping, DNA content measurement is one of the most routinely performed flow cytometry-based assays. These experiments require the use of DNA intercalating dyes, such as DAPI, Propidium Iodide, or Dye Cycle Violet, that when nested between DNA bases yield signals directly proportional to the amount of DNA within the nucleus [1]. Since DNA content changes predictably with the stage of the cell cycle, the stoichiometric nature of the dye interactions with nucleic acids makes it possible to determine the cell cycle profile of cultured cells (Figure 1). For example, cells that are rapidly dividing have greater S-phase content in contrast to cells arrested in the G0/G1 or G2/M replication checkpoints [2].
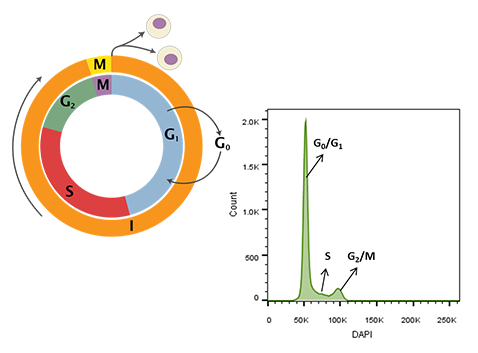
Figure 1. Cell cycle schematic (left) and DAPI-intensity histogram (right) obtained from analysis of DAPI stained single cells. DNA content doubles from G0/G1 to G2/M – the first peak in the histogram at ~50K median fluorescence intensity (MFI) is representative of G0/G1 whereas the second peak at ~ 100K MFI is representative of G2/M. Cells in S-phase have variable DNA content between that of cells in G0/G1 and G2/M during active DNA synthesis. Cell cycle schematic adapted from Richard Wheeler (Zephyris) 2006.
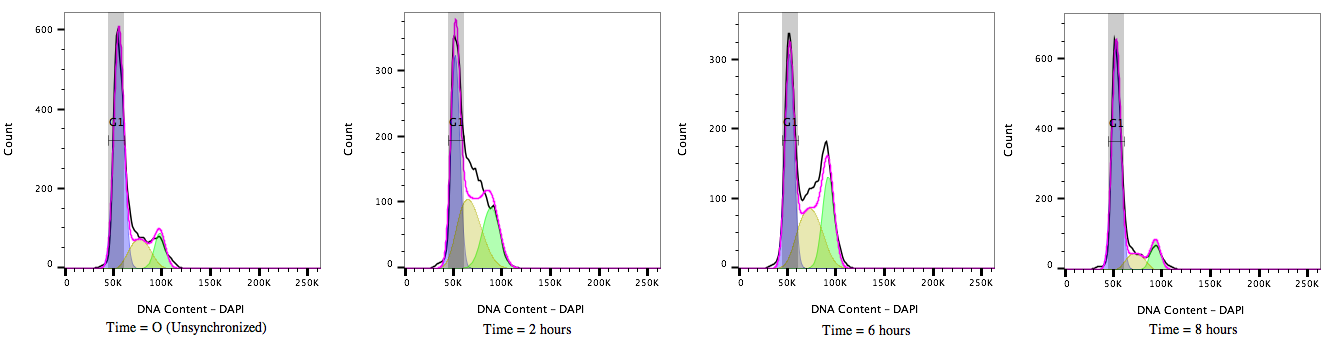
Figure 2: Cell cycle Progression of HEK 293T cells treated with a cell cycle modulating agent cells and stained with DAPI (4’, 6’-Diamidino-2-Phenylindole). Over time, from T = 0 to T= 8 hours, the cultures move through the cell cycle transitioning out of G1/0 (A) through S-phase (B) to G2M accumulation(C) and then cell division before resting again in the G1/0 phase (D).
Cancer cells and cell lines commonly have an irregular number of chromosomes (aneuploidy) caused by replication errors and the uneven distribution of genetic material during mitosis; in tumours, a greater degree of aneuploidy strongly correlates with aggressive hard to treat cancers [3, 4]. Ploidy determinations are important in characterizing tumours in the clinical setting, but are also useful in basic research when studying cancer cell drug responses in vitro.
When measuring ploidy, DNA content is described as a DNA Index (DI) or the ratio of the DNA content of the experimental sample to the DNA content of a corresponding normal control (diploid) population [1]. To properly calculate the DI, an internal DNA standard must be added to each sample to ensure accurate instrument setup and DNA measurements – chicken (CEN) and trout erythrocyte nuclei (TEN) are used for this purpose since their DNA content is known to be less than that of human cells. In fact, CENs have roughly one third the amount of genomic DNA per cell compared to humans [5] making them good internal standards as their DNA profiles will not overlap with those of the tested samples, see Figure 3.
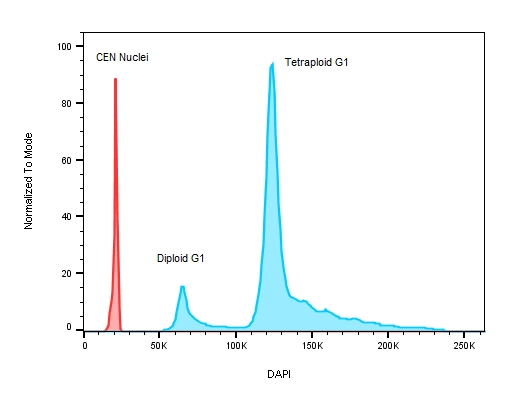
Figure 3: A plot of DNA content on a stem cell line with known replication defects causing the cells to have an abnormal DNA profile. The red peak on the histogram represents the internal control – chicken erythrocyte nuclei with lower roughly 3 x less DNA when compared to human ESC test samples where the ratio between CEN to the diploid G1 peak is 3.044 and 5.767 for the tetraploid G1.
Another useful control in the assessment of ploidy is non-proliferating human lymphocytes. In this scenario a patient’s own lymphocytes may be acquired alongside their tumour cells to measure any differences between the populations [6].
For accurate measurements, it is best to acquire data on isolated nuclei. Mitochondria multiply in response the increased energy demands of larger cells and have their own DNA and replication apparatus. As the mitochondrial burden in the cell increases this extra-nuclear DNA can increase background DNA fluorescence making ploidy measurements difficult [1].
[1] Darzynkiewicz, Z., Halicka, H. D., & Zhao, H. (2010). Analysis of Cellular DNA Content by Flow and Laser Scanning Cytometry. Advances in Experimental Medicine and Biology, 676, 137–147.
[2] Darzynkiewicz, Z., Traganos, F., Zhao, H., Halicka, H. D., & Li, J. (2011). Cytometry of DNA Replication and RNA Synthesis: Historical Perspective and Recent Advances Based on “Click Chemistry.” Cytometry. Part A : The Journal of the International Society for Analytical Cytology, 79(5), 328–337. http://doi.org/10.1002/cyto.a.21048
[3] Yildirim‐Assaf S, Coumbos A, Hopfenmüller W, Foss H, Stein H, Kühn W. The prognostic significance of determining DNA content in breast cancer by DNA image cytometry: the role of high grade aneuploidy in node negative breast cancer. Journal of Clinical Pathology. 2007;60(6):649-655. doi:10.1136/jcp.2005.035550.
[4] Rancés Blanco, Charles E. Rengifo, Mercedes Cedeño, Milagros Frómeta, and Enrique Rengifo, “Flow Cytometric Measurement of Aneuploid DNA Content Correlates with High S-Phase Fraction and Poor Prognosis in Patients with Non-Small-Cell Lung Cancer,” ISRN Biomarkers, vol. 2013, Article ID 354123, 8 pages, 2013. doi:10.1155/2013/354123
[5] Iversen, O. E. and Laerum, O. D. (1987), Trout and salmon erythrocytes and human leukocytes as internal standards for ploidy control in flow cytometry. Cytometry, 8: 190–196. doi:10.1002/cyto.990080212
[6] Benn, D. E. and Robinson, B. G. (1996), Peripheral blood mononuclear cells are a reliable internal standard for the flow cytometric DNA analysis of frozen malignant breast biopsies. Cytometry, 26: 161–165. doi:10.1002/(SICI)1097-0320(19960615)26:2<161::AID-CYTO10>3.0.CO;2-L
Dyes used for DNA content analysis will differ in their staining properties, and as such, specific protocols and certain advantages/disadvantages accompany the use of each specific dye. Cell fixation and permeabilization, achieved through various methods, is a requirement for cell cycle analysis with PI, 7-AAD, and DAPI. In addition, some dyes such as PI are not DNA-specific and will stain other double stranded nucleic acids such as RNA – therefore, when using PI, incubation of cells with RNase is necessary. In other circumstances dyes, like DAPI, are DNA-specific and obviate this problem (Darzynkiewicz et al.,2010).
Another important consideration when staining cells for this type of analysis is that cell concentrations must be consistent from sample to sample – thereby ensuring consistent saturation of DNA with the DNA binding dye. Otherwise, issues such as peak spreading and changes in G1 peak median fluorescence intensity from sample to sample arise – making experimental comparisons challenging. It is also important to note that the analysis of isolated nuclei may produce more accurate cell cycle characterization as opposed to analysis of whole cells, due to confounding mitochondrial DNA replication in actively proliferating cells (Darzynkiewicz et al.,2010).
Dyes like Hoechst33342, DRAQ5™ and Vybrant® DyeCycle allow for DNA content analysis within live samples making it possible to sort the cells by FACS for use in downstream assays on populations in specific cell cycle stages (Darzynkiewicz, 2011). Since these dyes are all membrane permeant no fixation/permeabilization step is required.
An important consideration to remember when anlayzing live stem cell populations is that drug efflux due to the high expression or P-glycoprotein and other multi-drug resistance pumps can occur which will prevent DNA saturation and the identification of a clear cell cycle profile. On the other hand, the dye efflux property of stem cells has been shown to be quite useful for identifying stem cells within a heterogeneous cell population through side population analysis.
2-Dimensional cell cycle analysis uses an additional parameter, such as proliferation, simultaneously with a stoichiometric DNA-binding dye to more accurately identify cells within different cell cycle stages (Darzynkiewicz and Zhao, 2014). For example, DAPI staining may be combined with fluorescently labelled-EdU or BrdU. In this assay, cells in S and G2 will not only have increased DAPI intensity relative to G1 cells but also fluorescence corresponding to EdU/BrdU S-phase incorporation (Figure 1).
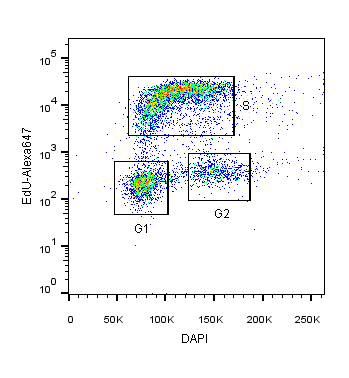
Figure 1: HEK293T cells co-stained with DAPI and EdU-Alexa 647 DNA binding dyes for the specific identification of cells in S-phase versus G1 or G2M cell cycle phases.
Staining for other markers associated with proliferation [such as; Ki67, phosphohistone H3, PCNA (proliferating cell nuclear antigen), Topoisomerase II and cyclin expression) may also be combined with a stoichiometric DNA-binding dye for increased accuracy and resolution in cell cycle analysis.
The FUCCI system of cell cycle tracking relies on the cell-cycle dependent oscillating expression of two proteins, Geminin and Cdt1 (Sakaue-Sawano et al., 2008). The expression of these proteins is regulated via cell cycle-dependent ubiquitin-mediated proteolysis. Cdt1 expression is high during G1, whereas Geminin expression is high during the S, G2 and M phases. As such, detection of the expression intensity of these proteins may be harnessed to determine the cell cycle profile of cell populations. When non-functional variants of these proteins were linked to different fluorescent indicator proteins and expressed in HeLa cells, the detection of fluorescent signals i.e. either from the presence of Ctd1 or Geminin, was correspondingly indicative of cell cycle stage (Sakaue-Sawano et al., 2008).
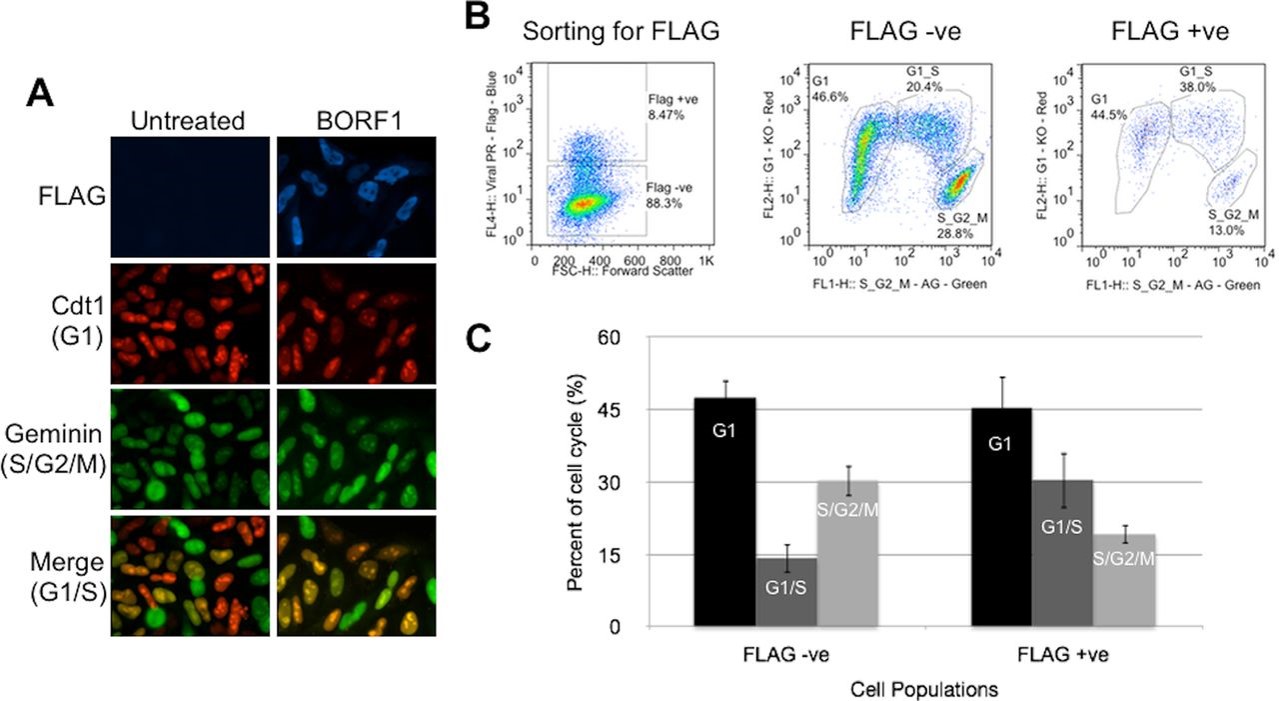
Figure 2: Screening cell cycle progression using Fucci cells. (A) Fucci cells were transfected with pMZS3F-BORF1 or left untreated and then stained for the FLAG tag on BORF1 and imaged for mKO2 (G1 cells; red), mAG (S/G2/M cells; green), and FLAG (blue). An overlay of red and green image layers revealed cells in G1/S, which display a yellow to orange color (bottom). (B) Fucci cells transfected with pMZS3F-BORF1 and stained for the FLAG tag on BORF1 were FACS sorted into FLAG-positive and FLAG-negative cells (left) and then further separated based on the expression of mKO2-Cdt1 and AG-geminin (middle and right). (C) Cell cycle stages determined from FACS analysis (as for panel B) were quantified for FLAG-positive (BORF1) and FLAG-negative cells from three independent experiments.
Patrick Paladino et al. J. Virol. 2014;88:4480-4492 (posted with permision from the American Society of Microbiology.)
Clonetech: Fluorescent Proteins and Reporters – Fucci Cell Cycle Vectors
ThermoFisher Scientific: Premo TM Fucci Cell Cycle Sensor (BacMam 2.0)
DeNovo Software: Book Chapter – DNA Analysis
Darzynkiewicz, Z., Halicka D. and Zhao, H. (2010) Analysis of cellular DNA content by flow and laser scanning cytometry. Adv Exp Med Biol. 676:137-147.
Darzynkiewicz, Z. (2011) Critical aspects in analysis of cellular DNA content. Curr Protoc Cytom. Apr. Unit 7.2.
Darzynkiewicz, Z. and Zhao, H. (February 2014) Cell Cycle Analysis by Flow Cytometry. In: eLS. John Wiley & Sons, Ltd:Chichester.DOI: 10.1002/9780470015902.a0002571.pub2
Sakaue-Sawano A., Kurokawa H., Morimura T., et al. (2008) Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3): 487-498.
Leers, M.P., Schutte, B., Theunissen, P.H., Ramaekers, F.C. and Nap, M. (1999) Heat pretreatment increases resolution in DNA flow cytometry of paraffin-embedded tumor tissue. Cytometry 35: 260-266.
Rabinovitch, P.S. (1994) in Darzynkiewicz, Z., Robinson, J.P. and Crissman, H.A. (eds.) Flow Cytometry. Methods in Cell Biology, 41, 264-296. Academic Press, Inc., San Diego.
Darzynkiewicz, Z., Robinson, J.P. and Crissman, H.A. (eds.) (1994) Flow Cytometry. Methods in Cell Biology, 41 & 42, Academic Press, Inc., San Diego.
Rabinovitch, P.S., Torres, R.M. and Engel, D. (1986) Simultaneous cell cycle analysis and two color surface immunofluorescence using 7-amino-actinomycin D and single laser excitation. J. Immunol. 136, 2769-2776.
Watson, J.V., Chambers, S.H. and Smith, P.J. (1987) A pragmatic approach to the analysis of DNA histograms with a definable G1 peak. Cytometry 8: 1-8.
Hedley, D.W., Friedlander, M.I., Taylor, I.W., Rugg, C.A. And Musgrove, E.A. (1983) Method for analysis of cellular DNA content of paraffin-embedded pathological material using flow cytometry.J. Histochem. Cytochem. 33: 1333-1335.
Vindelov, L.L., Christensen, I.J. and Nissen, N.I. (1983) A detergent-trypsin method for the preparation of nuclei for flow cytometric DNA analysis. Cytometry 3, 323-327.
